25% населения мира в возрастной группе от 55 лет и старше имеют семейную историю деменции. В большинстве случаев, семейные случаи данной патологии возникают вследствие наличия множества генетических дефектов, которые в совокупности значительно повышают риск развития деменции. Небольшая доля семей среди общей популяции имеет моногенные формы деменции с ранней манифестацией заболевания, которые обусловлены мутацией одного из генов деменции.
В этом обзоре авторы сфокусировали внимание на результатах и методах генетической диагностики болезни Альцгеймера (APP, PSEN1 и PSEN2 гены) с рассмотрением практических аспектов медико-генетического консультирования.
Введение
Когда у кого-то из родственников диагностируется деменция, клиницист часто может слышать вопрос: «У моей мамы деменция. Возможно, у меня тоже разовьется; могу ли я это проверить и предотвратить?»
Надо отметить, что отягченный анамнез по развитию деменции у родственников – необязательно означает наличие моногенной формы заболевания. Известно, что менделевские формы деменции крайне редки. К примеру, зарегистрированы лишь 500 семей с менделевскими формами болезни Альцгеймера. Таким образом, большинство людей с семейной историей заболевания не нуждаются в молекулярном генетическом тестировании и могут быть спокойны.
Как же идентифицировать то небольшое количество семей с повышенным риском развития деменции?
Менделевская форма заболевания и мультифакториальные заболевания
Каждая семья испытывает на себе влияние как окружающей среды, так и генетических факторов, поэтому семейные заболевания не всегда носят наследственный характер. Наиболее ярким примером деменции, вызванной факторами окружающей среды, является куру. Эта патология является инфекционной прионной болезнью, впервые зарегистрированной в 1950 году на папуасских островах Новой Гвинеи, где родственники употребляли в пищу умершего в погребальных ритуалах. Сначала все считали, что это заболевание генетической природы по причине его семейного характера, но результаты экспериментальных работ показали, что это, по сути, трансмиссивная спонгиформная энцефалопатия.
Однако, в подавляющем большинстве, известные нам факторы риска развития деменции генетической природы.
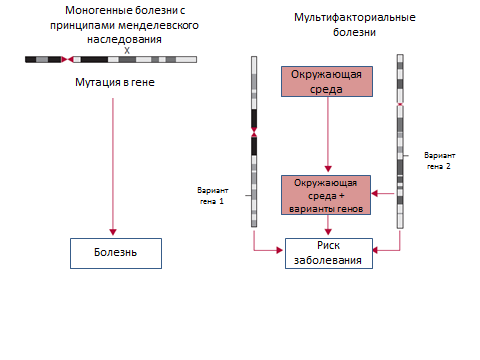
Генетические факторы могут вносить вклад в развитие семейной деменции двумя путями: приводя к менделевским формам заболевания (моногенным) или в качестве способствующего фактора мультифакториальных (полигенных) болезней (Рис.1).
Рисунок 1. Сравнение патогенеза моногенных и полигенных (мультифакториальных) заболеваний.
Клинические последствия менделевских форм деменции
Моногенные формы деменции с установленными генами-виновниками подчиняются аутосомно-доминантным принципам наследования и обладают высокой пенетрантностью, когда наблюдается высокий уровень заболеваемости в последующих поколениях. В этом случае очень полезным оказывается генетическое консультирование. Несмотря на наличие в генеалогическом древе лиц – носителей мутации, но не проявивших признаки заболевания в настоящее время, у этих людей в 95% и выше случаев развивается деменция. Точный риск развития заболевания зависит от возраста манифестации деменции в семье и пенетрантности гена, которая определяется возможностью развития фенотипа болезни у лица с мутацией в гене. Важно отметить, что лица, не несущие мутаций в геноме, имеют риск развития деменции такой же, как в общей популяции.
Принципы генетических исследований при деменции
Обдумывание необходимости генетического исследования включает в себя два шага. Первый шаг – детальный анализ истории семьи (наличие заболевания у родственников в нескольких поколениях) с дифференцировкой между моногенным типом наследования и мультифакториальными формами болезни. В данном аспекте семьям с аутосомно-доминантным типом наследования заболевания рекомендуется проведение генетического анализа. Второй шаг на пути к правильному решению – детальный анализ фенотипа заболевания с целью проведения адекватного потребностям генетического теста. К примеру, история психических нарушений как интегральный показатель фенотипа болезни, характерна для фронтотемпоральной деменции. А четкий учет возраста манифестации заболевания может служить вспомогательным фактором в определении вероятности развития болезни Альцгеймера в семьях, члены которых страдали данной патологией. Генетическое исследование наиболее правильно начинать с того члена семьи, который уже имеет признаки деменции.
Генетический аспект болезни Альцгеймера
Клинически, типичная форма болезни Альцгеймера характеризуется градуальным началом и быстрым прогрессированием в виде расстройств памяти и когнитивной дисфункции (по критериям 1984г. Тhe National Institute of Neurological and Communicative Disorders and Stroke–Alzheimer’s Disease and Related Disorders Association [NINCDS-ADRDA]). Эти диагностические критерии были пересмотрены с целью выявления форм болезни без нарушений памяти (с языковыми, зрительно-пространственными дисфункциями) и определения роли биомаркеров в диагностике (критерии 2011г. Тhe National Institute on Aging–Alzheimer’s Association [NIA-AA]).
Моногенные формы болезни Альцгеймера крайне редки: свыше 35 миллионов людей в мире страдают данной патологией, но генетические мутации установлены в случае 500 семей. Ключевыми элементами в анамнезе заболевания, помогающими отдифференцировать менделевское наследование от полигенного, являются: наследование болезни через поколения и ранний возраст манифестации болезни (таблица 1). Члены семей, отвечающие этим параметрам, несут в себе патогенетическую мутацию в одном из установленных для болезни генов. К примеру, Raux c коллегами, изучая когорту из 65 семей с ранней манифестацией заболевания (младше 60 лет) и наличием болезни в трех поколениях выявили: в 86% случаев – мутацию в гене-виновнике заболевания (78% - в виде изменения последовательности нуклеотидов, 8% - в виде патологической дупликации гена). Однако, семьи, отвечающие этим критериям, очень редко встречаются: распространенность в возрастной группе 41-60 лет около 5 на 100 000 человек.
Таблица 1. Вероятность наличия патогенетической мутации в одном из известных генов-виновников болезни Альцгеймера
| Возраст манифестации симптомов | Вероятность наличия мутации | |
|---|---|---|
| Наличие болезни в трех генерациях | < 60 лет | 86% |
| Наличие болезни у 2-ух и более родственников 1-ой линии | < 61 года | 68% |
| Наличие болезни у 2-ух и более родственников 1-ой линии | < 65 лет | 15% |
| Наличие болезни у 2-ух и более родственников 1-ой линии | старше 65 лет | < 1% |
Эта группа семей с аутосомно-доминантным наследованием патологии при манифестации болезни имеет ту же клиническую картину, что и другие формы болезни, однако развитие миоклонуса на ранних стадиях заболевания может быть диагностическим маркером моногенной болезни.
Известны три причинных гена болезни Альцгеймера: APP, PSEN1 и PSEN2. Надо отметить, что первый из них – ген APP на 21 хромосоме был обнаружен в 1987 году у лиц с трисомией 21 (синдромом Дауна), у которых развивается деменция со схожей гистопатологической картиной. Однако ни одной семьи с АРР-мутацией не было зарегистрировано до 1991 года. К тому времени было установлено, что главными гистопатологическими признаками болезни Альцгеймера является наличие амилоидных бляшек и нейрофибриллярных клубков. Дупликация АРР приводит к формированию бета-амилоида – главного компонента амилоидных бляшек. Эта находка легла в основу амилоидной гипотезы, предполагающей, что продукция и деградация бета-амилоида являются основными в патогенезе не только менделевской формы заболевания, но и болезни мультифакториальной, развившейся в общей популяции.
Впоследствии были выявлены еще два дополнительных гена-виновника: PSEN1 на хромосоме 14 и PSEN2 на хромосоме 1. В обоих случаях, мутации приводили к гиперпродукции бета-амилоида или, в некоторых случаях, к изменению соотношения изоформ бета-амилоида1-42 к бета-амилоиду1-40 (рис.1).
Рисунок 1. Амилоидная гипотеза и патогенез болезни Альцгеймера.
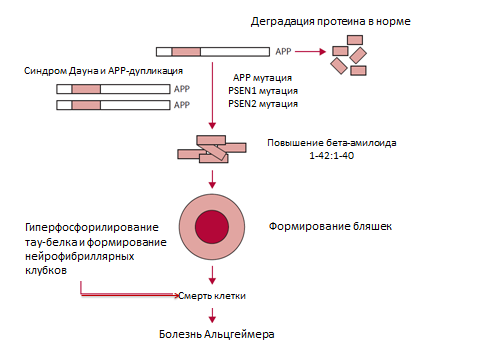
Так сформировалась амилоидная гипотеза, являющаяся доминирующей парадигмой в изучении данного заболевания. Хотя патогенез заболевания в общей популяции, вероятно, более сложный и многофакторный.
Как отмечалось ранее, 86% семей с ранней манифестацией деменции (в возрасте ранее 60 лет) в трех и более поколениях имеют мутации в генах APP, PSEN1 или PSEN2. Мутации PSEN1 – наиболее частая причина, встречающаяся у 60% семей с менделевской формой болезни. Около 15% семей имеют мутацию в виде изменения нуклеотидной последовательности АРР, а в 8% случаев встречается дупликация АРР. Мутации PSEN2 достаточно редка: установлена лишь у 22 семей.
Таким образом, при необходимости, в первую очередь проводится скрининг на наличие мутации PSEN1. Существуют также клинические ключи к выбору приоритетного генетического анализа. Так, наличие болезни Альцгеймера и спастического парапареза более характерно при наличии PSEN1 мутации. В то время как АРР мутация часто приводит к церебральной амилоидной ангиопатии с развитием церебральных кровоизлияний.
Практические аспекты медико-генетического консультирования
Генетическое консультирование обычно проводится у лиц с уже развившимся заболеванием и в целях прогноза. В обоих случаях эта процедура бывает полезной. Разумеется, генетическое исследование у лиц с развившейся болезнью, не влияет на клиническое ведение пациента, но может служить хорошим помощником в постановке диагноза, когда он не столь очевиден, а также, в случае выявления моногенной болезни, быть поводом для консультации всех членов семьи.
Важно, что необходима очная консультация генетика с выяснение всех «точек над и», когда дело касается проведения генетических анализов для определения прогноза развития болезни в будущем. И главное в этом аспекте – понимание, что никаких превентивных мер лечения этой патологии не существует. В таких случаях, члены семьи желают провести генетическое обследование по трем причинам: вопрос функции памяти, планирование будущей жизни и планирование деторождения. Отмечено, что в случае получения положительных результатов существует повышенный риск суицидов, поэтому не рекомендуется проводить обследование у пациентов с психологическими и психиатрическими проблемами.
Заключение
К счастью, моногенные формы деменции редко встречаются. Родственники пациентов с развившейся болезнью, имеют риск развития деменции в течение всей жизни около 20% по сравнению с 10% в общей популяции. Однако, небольшая когорта семей с аутосомно-доминантными формами наследования имеют мутацию одного из генов-виновников. Каждый ребенок пациента с моногенным заболеванием имеет 50% шанс иметь ту же мутацию и при наличии её – подвергается 95% риску развития болезни в течение жизни. Около 50% родственников в таких семьях после детальной консультации с генетиком и лечащим врачом отказываются от генетического обследования на предмет выявления генов-виновников еще не развившегося заболевания.
Источник: Clement T Loy, Prof Peter R Schofield, Anne M Turner. Genetics of dementia. The Lancet. Published online August 6, 2013 http://dx.doi.org/10.1016/S0140-6736(13)60630-3